Aktuelles

Immobilieninvestition mit Weitblick: Wie die Dr. Barbara Mez-Starck Stiftung nachhaltigen Mehrwert schafft
Die Entscheidung der Dr. Barbara Mez-Starck Stiftung, in den Bau einer Immobilie zu investieren, markiert einen Wendepunkt in der Geschichte der Stiftung und unterstreicht deren Engagement für die Förderung der Wissenschaft und Bildung. Diese Investition geht weit über den finanziellen Aspekt hinaus und trägt auf vielfältige Weise zur Erfüllung des Stiftungszwecks bei.
Förderung von Wissenschaft und Bildung
Die Stiftung hat mit dem Bau des Mez-Starck-Hauses nicht nur eine sichere Kapitalanlage getätigt, sondern auch einen Ort geschaffen, der die Forschung und Bildung auf mehreren Ebenen unterstützt. Senator Werner Braun betont die Bedeutung dieser Investition: „Mit der Investition in eine nachhaltige Immobilie am Campus der Universität Ulm tätigen wir nicht nur eine kluge Vermögensanlage, sondern fördern gleichzeitig den wissenschaftlichen Fortschritt."
Das Gebäude bietet dem Institut für Theoretische Chemie und dessen Arbeitsgruppe Chemical Information Systems moderne und effiziente Arbeitsumfelder, die ihre Forschung im Bereich der Naturwissenschaften positiv beeinflussen werden. „Dieses Gebäude wird nicht nur die Arbeitsbedingungen für uns Wissenschaftlerinnen und Wissenschaftler verbessern, sondern auch ein Anziehungspunkt für junge Talente sein", unterstreicht Prof. Dr. Axel Groß die Bedeutung dieser Einrichtung für die wissenschaftliche Gemeinschaft.
Nachhaltige und langfristige Finanzsicherung
Durch die Vermietung des Gebäudes an die Universität Ulm sichert die Stiftung nicht nur langfristige Einnahmen, sondern stärkt auch die Bindung zwischen der Stiftung und der Universität. Dies unterstreicht das Engagement der Stiftung, langfristig zum Wissenschaftsstandort Ulm beizutragen.
Ein architektonisches Konzept mit Weitblick
Das von Architekt Stefan Rapp entworfene Mez-Starck-Haus am Oberen Eselsberg ist ein Paradebeispiel für zukunftsorientiertes Bauen. „Unser Ziel war es, ein Gebäude zu schaffen, das nicht nur funktional und ästhetisch ansprechend ist, sondern auch den ökologischen Aspekten Rechnung trägt", erklärt Architekt Rapp. Durch die Verwendung von Holz und Beton in einer Hybridbauweise wurde eine nachhaltige und ressourcenschonende Architektur realisiert, die den zirkulären Gedanken in den Vordergrund stellt und somit einen bedeutenden Beitrag zur Schonung der Umwelt leistet.
Eine beispielhafte Investition für Stiftungen
Die Investition der Dr. Barbara Mez-Starck Stiftung in den Bau einer Immobilie für die Universität Ulm ist ein beispielhaftes Modell für die Verbindung von ökonomischer Weitsicht und ökologischer Verantwortung. Das Mez-Starck-Haus steht nicht nur für nachhaltige Architektur und die Förderung der Wissenschaft, sondern auch für die visionäre Strategie einer Stiftung, die es versteht, ihren Stiftungszweck auf innovativen Wegen zu erfüllen. Damit setzt die Dr. Barbara Mez-Starck Stiftung Maßstäbe für zukunftsorientiertes Engagement und unterstreicht die Bedeutung von Investitionen, die sowohl finanzielle Sicherheit als auch gesellschaftlichen Mehrwert schaffen.

Erfolgschancen auf Exzellenzcluster steigen für die Uni Ulm
Chem4Quant hat sich gegen weitere Bewerber durchgesetzt
Die Universität Ulm hat in einer strategischen Partnerschaft mit dem renommierten Karlsruher Institut für Technologie (KIT) sowie der Universität Stuttgart einen bedeutenden Schritt vorwärts gemacht, indem sie sich gemeinsam um einen prestigeträchtigen Exzellenzcluster beworben haben. Diese Bewerbung markiert einen wichtigen Meilenstein in ihrem Bestreben, ihre Forschungskapazitäten und -kompetenzen weiter auszubauen und zu stärken.
Im Rahmen dieses ambitionierten Projekts hat das Konsortium nun eine signifikante Hürde überwunden: Unter der Vielzahl von Einreichungen, genauer gesagt 143 Antragsskizzen, hat es die "Chem4Quant"-Initiative der Universität Ulm geschafft, sich in dem hochkompetitiven Auswahlprozess durchzusetzen. Lediglich 41 der eingereichten Skizzen wurden für die nächste Phase der Antragstellung – die Einreichung eines Vollantrags – ausgewählt. Dieser Erfolg unterstreicht nicht nur die hohe Qualität und Innovationskraft der vorgeschlagenen Forschungsideen, sondern positioniert die "Chem4Quant"-Initiative auch hervorragend im Rennen um die begehrte Förderung. Sollte der Vollantrag erfolgreich sein, wird die Initiative Teil eines exklusiven Kreises von 98 Exzellenzclustern sein, die eine Förderung beantragt haben, einschließlich der 57 Cluster, die bereits seit 2019 unterstützt werden.
Im Zentrum des wissenschaftlichen Vorstoßes in unentdeckte Bereiche der Quantentechnologie steht der Exzellenzcluster „Chem4Quant“. Hier werden mit mikroskopischer Präzision Materialstrukturen entwickelt, die als Fundament für die fortschrittlichsten Quantentechnologien der Zukunft dienen sollen. Experten aus den Feldern der Computerwissenschaften, Materialkunde, Physik und Chemie bündeln ihr fachspezifisches Know-how, um in interdisziplinärer Kollaboration neue Erkenntnisse zu generieren.
„Chem4Quant“ besitzt ein starkes Netzwerk aus etablierten Kooperationen und somit eine Expertise im Bereich der molekularen Quantensysteme, die weltweit einmalig ist. Chemisch gezielt definierbare Quantenarchitekturen dienen beispielsweise als Grundlage für ein zukünftiges Quanteninternet. Unter Nutzung einer chemischen Basis werden Qubit-Materialien so aufgebaut, dass sie den speziellen quantentechnologischen Anforderungen entsprechen.
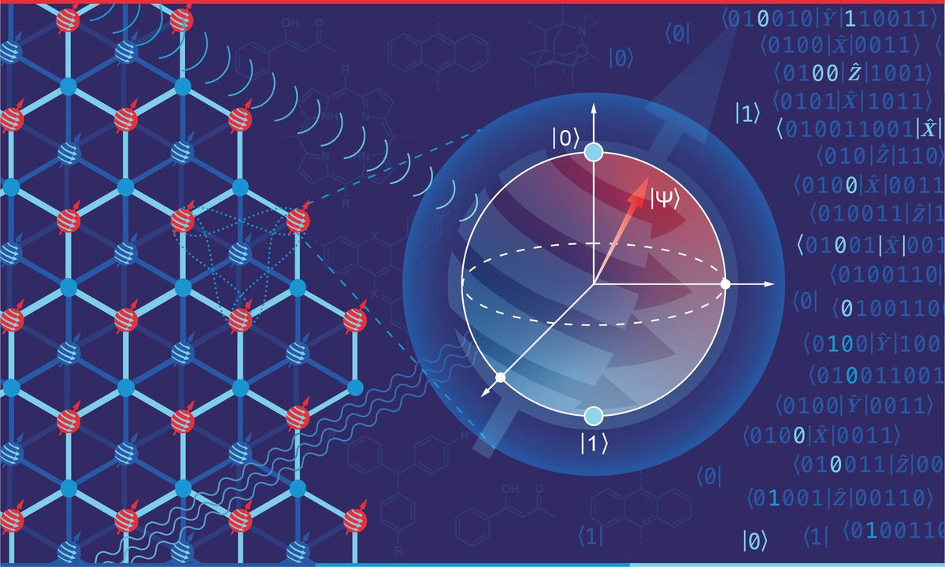
Dieser Ansatz ist deshalb so besonders, weil viele der aktuell genutzten chemischen Plattformen in Hinsicht auf genaue Definition, Skalierbarkeit, Positionierungsmöglichkeiten sowie Ausgleich von Fehlern limitiert sind. Es wäre erstmals eine Positionierung von elektrischen oder photonischen Modulen möglich, die unterhalb des Nanometerbereichs liegt. Durch die Erforschung innovativer Qubit-Materialien können erste Elemente für das künftige Quanteninternet verwirklicht werden.
Diese Vorreiterrolle in der Nanotechnologie markiert einen Wendepunkt hin zu einer Ära, in der die Quantenkommunikation die Grenzen theoretischer Modelle überwindet und praktisch implementierbar wird. Das ambitionierte Forschungsprojekt von "Chem4Quant" ist darauf ausgerichtet, ein robustes Fundament für die Realisierung einer skalierbaren und präzise steuerbaren Quanteninfrastruktur zu schaffen. Solch eine Infrastruktur ist für die Integration von Quantentechnologien in kommerzielle Anwendungen unerlässlich. Die Überwindung aktueller Einschränkungen in der Präzisionschemie eröffnet nicht nur innovative Perspektiven für den Quantencomputing-Sektor, sondern ebnet auch den Weg für die Entwicklung von Quantenkommunikationssystemen, die potenziell einschneidende Auswirkungen auf unsere Informationsgesellschaft haben könnten. Jede wissenschaftliche Errungenschaft, die im Rahmen von "Chem4Quant" erzielt wird, leistet somit einen essenziellen Beitrag zur Transformation der Quantentechnologie von einer faszinierenden theoretischen Möglichkeit in eine praktisch umsetzbare, fortschrittliche Technologie.
Mit den Initiativen Chem4Quant und POLiS positioniert sich die Universität Ulm nun mit zwei vielversprechenden Anträgen im Wettbewerb um die begehrten Exzellenzcluster. Das Projekt POLiS (Post-Lithium Energy Storage) wird bereits seit 2019 gefördert und steht exemplarisch für die herausragende Forschung an der Universität. Im Zentrum dieser Initiative steht das renommierte Institut für Theoretische Chemie der Universität Ulm, das sich unter anderem durch die Unterstützung der Dr. Barbara Mez-Starck-Stiftung auszeichnet. Prof. Dr. Axel Groß, der Direktor des Instituts, nimmt als einer der führenden Sprecher des Clusters eine zentrale Rolle ein und trägt entscheidend zur Erstellung des Fortsetzungsantrags bei, der in Kooperation mit dem Karlsruher Institut für Technologie (KIT) und der Justus-Liebig-Universität Gießen eingereicht wird.
Das von den beteiligten Wissenschaftlerinnen und Wissenschaftlern gezeigte Engagement und der Forschungseifer verleihen dem Institut eine vitale Rolle innerhalb dieses Netzwerks. Gleichzeitig spiegeln sie die tiefe Verwurzelung des Instituts in den Prinzipien akademischer Exzellenz wider. Durch die Projekte Chem4Quant und POLiS eröffnen sich zudem wertvolle Synergieeffekte, insbesondere in der materialorientierten Grundlagenforschung, die das Potential haben, die Forschungslandschaft nachhaltig zu prägen.
Die Bewilligung dieser beiden Clusteranträge würde der Universität Ulm die Möglichkeit eröffnen, im Rahmen der Exzellenzstrategie des Bundes und der Länder einen weiteren, signifikanten Schritt zu gehen: die Beantragung der Förderlinie „Exzellenzuniversität“. Eine solche Entwicklung könnte die Sichtbarkeit und Anerkennung der Forschungsaktivitäten der Universität Ulm erheblich steigern. Die Anerkennung als Exzellenzuniversität wäre nicht nur eine Bestätigung für die bisher geleistete Arbeit, sondern auch ein wichtiger Katalysator für zukünftige Forschungsinitiativen. Sie würde es der Universität ermöglichen, auf nationaler und internationaler Ebene noch stärker als führende Forschungseinrichtung wahrgenommen zu werden und ihre Position an der Spitze wissenschaftlicher Innovation und Exzellenz weiter zu festigen.
Entscheidung für zukünftige Exzellenzcluster erst in 2025
Das Förderprogramm Exzellenzcluster ist Bestandteil der Exzellenzstrategie von Bund und Ländern und wird von der Deutschen Forschungsgemeinschaft (DFG) realisiert. Die ausgewählten Exzellenzcluster erhalten eine jährliche Förderung von drei bis zehn Millionen Euro für jeweils sieben Jahre. Es sind maximal zwei Förderperioden von somit insgesamt 14 Jahren möglich. Ende Mai 2015 wird die finale Förderentscheidung über die künftigen Exzellenzcluster getroffen.

Interview mit Prof. Dr. Melanie Schnell
Hallo Frau Prof. Dr. Melanie Schnell, schön dass Sie sich etwas Zeit für ein Interview mit uns nehmen. Wenn man Sie googelt bekommt man schnell einen ersten Eindruck, an was für Themen Sie in ihrem Fachgebiet bis heute bereits geforscht haben und vor allem, wo Sie überall herumgekommen sind in der Welt. Wir sind schwer beeindruckt und finden das Thema ihrer Forschung sehr spannend, auch wenn wir technisch gesehen recht wenig davon verstehen, müssen wir zugeben.
Wann haben Sie denn in Ihrem Leben angefangen sich für die Forschung – und speziell für die Physik und die Chemie – zu interessieren?
Das Interesse an Forschung ist bei mir bereits zu Beginn des Studiums aufgekommen. Ich fand und finde es faszinierend, mir grundlegende Kenntnisse anzueignen und dann daran zu arbeiten, diese weiter zu entwickeln.
Ich wuchs dörflich und recht naturnah auf. Meine Großmutter hatte einen sehr großen Gemüsegarten. Schon früh fragte ich mich, warum beispielsweise manche Pflanzen aus „Zwiebeln“, andere aus „Bohnen“ und wieder andere aus kleinen Samen wuchsen. Mit dieser frühen Neugier für solche eher biologischen Zusammenhänge entdeckte ich in der Schule mein Interesse für die Chemie, bis ich dann im Studium bei der Physikalischen Chemie ankam – für mich ideal: Physikalische Methoden zu entwickeln, um chemische Zusammenhänge zu verstehen. Auch hier kam und kommt mir wieder meine Kindheit entgegen. Mein Vater baute zu Hause vieles selbst – und wir Kinder durften und (manchmal) mussten helfen. So wurde ich früh an Werkzeuge und handwerkliches Arbeiten herangeführt – eine wichtige Grundlage für unsere Laborarbeiten.
Erklären Sie uns doch bitte kurz, für was Ihr Forschungsbereich steht und wo Ihre Ergebnisse überall genutzt werden können?
Kurzfassen ist nicht meine Stärke, daher hier eine etwas ausführlichere Kurzversion.
Wir entwickeln und nutzen spektroskopische Methoden, um Molekülverbindungen mit großer Genauigkeit untersuchen zu können. Unser Forschungsinteresse konzentriert sich da jeweils auf die molekulare Ebene – wir wollen z.B. verstehen, wie Moleküle im Detail miteinander wechselwirken und damit, wie schon kleinste Veränderungen an diesen Molekülen zu anderen Wechselwirkungen führen können. Und diese Wechselwirkungen sind es schlussendlich, die die Funktion und damit Wirksamkeit von Molekülverbindungen bestimmt. Zum Beispiel können selbst relativ kleine Moleküle ihre bevorzugte Struktur ändern, je nachdem, ob sie sich in Wasser oder Ethanol, d.h. Alkohol, als Lösungsmittel befinden. Dieses gilt es zu verstehen.
Ein zweiter Schwerpunkt unserer Forschung liegt auf den Untersuchungen chiraler Moleküle. Diese Moleküle kommen in zwei Formen vor, den Enantiomeren, die wie unsere beiden Hände Spiegelbildcharakter haben, d.h. nicht einfach ineinander überführt werden können. Während diese beiden Enantiomere identische physikalische Eigenschaften wie Siedepunkte und Schmelzpunkte aufweisen, können sie hochgradig verschiedene biochemische Eigenschaften haben. Dieses zeigt sich beispielsweise in unterschiedlichen Wirksamkeiten chiraler Wirkstoffmoleküle wie Ibuprofen: Während das eine Enantiomer, d.h. die eine Händigkeit, des Ibuprofens die bekannte schmerzstillende und entzündungshemmende Wirkung hat, ist das andere Enantiomer zunächst weitestgehend unwirksam und wird schließlich im Körper durch ein Enzym in die wirksame Komponente umgewandelt – eine interessante Langzeitwirkung. Diese Unterschiede in der biochemischen und medizinischen Wirksamkeit gilt es auf der Molekülebene zu verstehen. Darüber hinaus entwickeln wir Methoden, mit denen wir chirale Molekülverbindungen identifizieren und unterscheiden können.
Unser dritter Forschungsbereich bringt uns weg von der Erde ins Weltall. Wir charakterisieren Molekülverbindungen, die auch eine Rolle in der Chemie des interstellaren Raums spielen können. Dieser Raum zwischen den Sternen ist nicht leer, sondern gefüllt mit Molekülwolken, die sich unter bestimmten Umständen weiter verdichten und dann der Beginn eines neuen Sterns werden können. Das Inventar solcher Molekülwolken wird mit Teleskopen untersucht. Diese Teleskope können die Molekülfingerabdrücke, die die Molekülverbindungen nach Anregung durch Sternenlicht aussenden, einsammeln. Um diesen spektralen Fingerabdrücken spezielle Molekülverbindungen zuordnen zu können, untersuchen wir geeignete Molekülverbindungen im Labor und stellen diese Informationen Radioastronominnen und -astronomen, die die Teleskope betreiben, für ihre Analyse zur Verfügung. So kann man nach und nach das große Puzzle, welche Molekülverbindungen im Weltall vorkommen und wie diese gebildet wurden und ggf. weiterreagieren, vervollständigen.
Sie sind seit einigen Jahren Professorin an der Christian-Albrechts-Universität zu Kiel und Leitende Wissenschaftlerin bei DESY in der Forschungsgruppe „Spektroskopie molekularer Prozesse“. Woran forschen Sie denn aktuell im Speziellen? Auf was darf man gespannt sein?
Allgemeine Beispiele unserer Forschung hatte ich ja gerade bereits genannt. Ganz aktuell beschäftigen wir uns zum Beispiel damit, die Enantiomere chiraler Moleküle nicht nur zu unterscheiden, sondern auch zu trennen, so dass wir dann sehr kontrollierte Untersuchungen an ihnen vornehmen können. Unsere Methode ist insbesondere für die Molekülsysteme interessant, deren Enantiomere nicht einfach auf anderen Wegen getrennt werden können. Hier kommen wir ins Spiel.
Was war für Sie der Anlass, sich zukünftig bei der Dr. Barbara Mez-Starck-Stiftung mit einzubringen? Wie haben Sie von der Stiftung erfahren?
Ich kenne die Dr. Barbara Mez-Starck-Stiftung schon seit vielen Jahren, in erster Linie über die Mogadoc-Datenbank, die wir bereits während meiner Promotionszeit an der Universität Hannover verwendet haben, um spektroskopische Informationen zu bestimmten Molekülverbindungen schnell zur Hand zu haben. In meinem Forschungsbereich der Molekülspektroskopie ist die Dr. Barbara Mez-Starck-Stiftung auch über den internationalen Dr. Barbara Mez-Starck-Forschungspreis über alle Ländergrenzen hinaus bekannt, und ich fühlte mich sehr geehrt, als ich 2021 mit eben diesem Preis ausgezeichnet wurde.
Sie haben für ihre Forschungsergebnisse bereits zahlreiche Preise und Auszeichnungen gewonnen – unter anderem 2013 den Helene-Lange-Preis – der ausschließlich Nachwuchswissenschaftlerinnen in den MINT-Fächern und deren überragende Leistungen Tribut zollt und sie für ihre Leistungen ehrt. Wie sehen Sie das nach wie vor ungleiche Verhältnis der Geschlechter in den Wissenschaften?
Zunächst einmal ist es mir ein generelles Anliegen, junge Leute für die Naturwissenschaften zu begeistern, z.B. mit Vorträgen, durch SchülerInnenpraktika in der Forschungsgruppe oder bei Besuchen von Schülerinnen und Schüler. Darüber hinaus bin ich fest davon überzeugt, dass Rollenvorbilder sehr wichtig sind, um sich bestimmte Berufs- und Entwicklungswege besser vorstellen zu können und auch zuzutrauen. Hier versuche ich sehr aufmerksam zu sein, z.B. indem ich bei Konferenzen, die wir planen, besonders darauf achte, einen ausgeglichenen Anteil an Wissenschaftlerinnen einzuladen. Dieses erhöht zum einen die Sichtbarkeit der einzelnen Personen, aber auch die Sichtbarkeit von Wissenschaftlerinnen im Allgemeinen. Als ich Ende der 1990er Chemie studierte, gab es an meiner Universität in diesem Fach keine Professorin. Zunächst fiel mir dieses nicht wirklich auf. Erst nach einem Uniwechsel merkte ich, dass es für mich motivierend war, auch mit Professorinnen zusammen zu arbeiten – Rollenvorbildern eben. Vielfalt ist wichtig, so bleiben alle offen für anderen Ansichten und Ideen. Gerade in Zeiten, in denen einige Leute in öffentlichen Positionen vermeintlich einfache Lösungen versprechen, wird dieses immer wichtiger.
Als letztes interessiert uns noch brennend: Sind sie auch auf Social-Media-Kanälen aktiv und wenn ja, mit welcher Intention und auf welchen Plattformen?
Nein, wir sind als Forschungsgruppe nicht auf Social-Media-Kanälen unterwegs. Wir haben eine aktive Website und betreiben viel outreach, z.B. mit Schulen, wie vorher erwähnt, aber keine Social-Media-Aktivitäten.
Wir bedanken uns herzlich für Ihre Zeit und freuen uns in Zukunft mehr von Ihnen aus dem Umfeld der Dr. Barbara-Mez-Starck-Stiftung zu hören.

Wie die Entdeckungen des theoretischen Wissenschaftlers Dr. Sotoudehs die Batteriewelt verändern.
Ein Interview zur Zukunft der Energiespeicher
Lasse Martinsen: Herr Dr. Mohsen Sotoudeh, Sie als führender theoretischer Wissenschaftler im Bereich Biochemie erzählen uns heute etwas über Ihre Arbeit und die Zukunft der Batterietechnologie. Könnten Sie uns zunächst ein wenig über Ihren Werdegang und Ihre aktuelle Tätigkeit erzählen?
Dr. Mohsen Sotoudeh: Natürlich, Herr Martinsen, und vielen Dank für die Einladung. Ich arbeite seit über einem Jahrzehnt im Bereich der Materialwissenschaft, der theoretischen Physik und Chemie, mit einem besonderen Fokus auf Batterietechnologien und elektrochemischen Speichersystemen. Derzeit verfasse ich meine Habilitation an der Universität Ulm, wo ich mich auf die Entwicklung neuer Materialien für leistungsfähigere und nachhaltigere Batterien konzentriere.
Lasse Martinsen: Das klingt nach einer spannenden und zukunftsweisenden Arbeit. Können Sie uns ein Beispiel für eine Entdeckung geben, auf die Sie besonders stolz sind?
Dr. Mohsen Sotoudeh: Einer der Höhepunkte meiner Karriere war die Entwicklung eines sogenannten Deskriptors für die Ionenmobilität, der eine starke Korrelation zwischen der Aktivierungsenergie für Diffusionsereignisse und der Ionenleitfähigkeit von Materialien zeigt. Diese Arbeit ermöglichte es uns, komplexe Eigenschaften wie die Ionenmigration durch einfache physikalische Parameter wie Elektronegativität und Ionenradien zu verstehen und vorherzusagen. Das Besondere daran war, dass wir durch diesen Ansatz in der Lage waren, Materialien zu identifizieren, die für den Einsatz in Batterien besonders geeignet sind, ohne sie tatsächlich herstellen und testen zu müssen. Diese Entdeckung, die wir durch die Einführung des Konzepts der Bindungscharakteristik mittels Elektronegativität erreicht haben, stellte einen großen Schritt in der Materialwissenschaft dar und wurde durch eine lineare Skalierungsbeziehung bestätigt. Auf diesen Erfolg bin ich stolz, da es den Weg für schnelle und effiziente Entdeckungen neuer Materialien für Batterieanwendungen ebnet.
Lasse Martinsen: Sie erwähnen, dass wissenschaftliche Entdeckungen Zeit brauchen, um in praktische Anwendungen überführt zu werden. Wie gehen Sie mit der Ungeduld der Gesellschaft um, die oft nach sofortigen Lösungen sucht?
Dr. Mohsen Sotoudeh: Das ist eine Herausforderung, mit der viele Wissenschaftler konfrontiert sind. Es ist wichtig, eine Balance zu finden zwischen der Begeisterung für das Potenzial neuer Entdeckungen und der Geduld, die notwendig ist, um diese Entdeckungen sorgfältig zu validieren und weiterzuentwickeln. Wir bemühen uns, transparent zu kommunizieren, was machbar ist und was noch Zeit braucht, um Missverständnisse zu vermeiden.
Lasse Martinsen: Neben Ihrer Forschung scheinen Sie auch einen starken Fokus auf die Zusammenarbeit zu legen, sowohl innerhalb Ihrer Disziplin als auch darüber hinaus. Können Sie uns mehr darüber erzählen?
Dr. Mohsen Sotoudeh: Ja, die interdisziplinäre Zusammenarbeit ist entscheidend. In meiner Arbeit begegne ich Kollegen aus der Physik, Chemie, Materialwissenschaft und sogar der Ingenieurwissenschaft. Diese Vielfalt an Perspektiven bereichert unsere Forschung enorm. Darüber hinaus lege ich großen Wert auf die Zusammenarbeit mit der nächsten Generation von Wissenschaftlern. Sie bringen frische Ideen und neue Ansätze, die für die Lösung der komplexen Herausforderungen, vor denen wir stehen, unerlässlich sind.
Lasse Martinsen: Sie sehen Sie die Wichtigkeit von Transparenz und Offenheit in der Wissenschaft und wie wirkt sich das auf Ihre Arbeit aus?
Dr. Mohsen Sotoudeh: Transparenz fördert nicht nur das Vertrauen in die Wissenschaft, sondern beschleunigt auch den Erkenntnisgewinn. Durch das Teilen von Daten, Methoden und Ergebnissen können wir Doppelarbeit vermeiden und auf den Erkenntnissen anderer aufbauen. Dies ist besonders in meinem Feld der Batterieforschung von Bedeutung, wo ein schneller Fortschritt dringend benötigt wird, um den globalen Energieherausforderungen zu begegnen.
Lasse Martinsen: Was würden Sie Studierenden oder jungen Forschenden raten, die eine Karriere in Ihrem Forschungsbereich anstreben?
Dr. Mohsen Sotoudeh: Ich würde ihnen raten, neugierig zu bleiben und ihre Leidenschaft für die Wissenschaft zu pflegen. Es ist wichtig, sich nicht nur auf ein enges Fachgebiet zu beschränken, sondern auch interdisziplinäre Kompetenzen zu entwickeln. Die Welt der Batterietechnologie und Materialwissenschaft ist unglaublich vielfältig und bietet zahlreiche Möglichkeiten für innovative Forschung. Zudem sollten sie die Bedeutung von Kommunikation und Zusammenarbeit nicht unterschätzen. Die Fähigkeit, Ideen klar zu vermitteln und mit anderen Disziplinen zusammenzuarbeiten, ist entscheidend für den wissenschaftlichen Fortschritt.
Lasse Martinsen: Haben Sie als Forscher nicht auch Bedenken hinsichtlich des Diebstahls Ihrer Ideen, wenn Sie so sehr auf Transparenz setzen?
Dr. Mohsen Sotoudeh: Ich bin der Überzeugung, dass Transparenz tatsächlich der Schlüssel zum Schutz und zur klaren Zuordnung von Ideen ist. Je offener und freier Ideen geteilt werden, desto eindeutiger lässt sich nachverfolgen, wer den ursprünglichen Ansatz eingebracht hat.
Lasse Martinsen: Sie haben sich vor einigen Jahren speziell für die Universität Ulm entschieden. Was war der ausschlaggebende Grund für Ihren Wechsel hierhin, insbesondere im Hinblick auf Ihre Forschung in der Batterietechnologie?
Dr. Mohsen Sotoudeh: Der Hauptgrund für meinen Wechsel an die Universität Ulm war die exzellente Forschungsinfrastruktur und das starke Engagement der Universität in der Batterietechnologie und Materialwissenschaft. Ulm ist bekannt für seine führende Rolle in diesen Bereichen, unterstützt durch die enge Zusammenarbeit mit Industriepartnern und anderen Forschungseinrichtungen. Insbesondere die Förderung durch die Deutsche Forschungsgemeinschaft (DFG), die Mez Starck Stiftung und die Beteiligung an verschiedenen interdisziplinären und internationalen Projekten bieten ideale Voraussetzungen für zukunftsweisende Forschung. Hinzu kam die Möglichkeit, mit führenden Experten auf dem Gebiet der Batterieforschung zusammenzuarbeiten, was für meine Entscheidung, nach Ulm zu kommen, ausschlaggebend war. Die Kombination aus hervorragender Forschungsumgebung, Förderung innovativer Projekte und der offenen, kollaborativen Atmosphäre hat Ulm zum idealen Ort für meine wissenschaftliche Laufbahn und Weiterentwicklung gemacht.
Lasse Martinsen: Wie sehen Sie die Rolle Deutschlands in der globalen Batterieforschung und -technologie?
Dr. Mohsen Sotoudeh: Deutschland hat eine starke Tradition in der Forschung und Entwicklung, insbesondere in den Ingenieurwissenschaften und der Chemie, was eine solide Grundlage für Innovationen in der Batterietechnologie bietet. Allerdings gibt es im internationalen Vergleich, besonders mit Ländern wie China oder den USA, die sehr stark in die Batterieforschung investieren, noch Aufholbedarf. Deutschland hat das Potenzial, eine führende Rolle einzunehmen, insbesondere durch die Förderung von Forschungsinitiativen und die Zusammenarbeit zwischen Universitäten, Stiftungen, Forschungsinstituten und der Industrie. Die jüngsten Investitionen in Batterieforschungszentren und die Fokussierung auf die Entwicklung nachhaltiger Batterietechnologien sind positive Schritte in diese Richtung.
Lasse Martinsen: Welche spezifischen Herausforderungen sehen Sie in der Entwicklung der nächsten Generation von Batterien?
Dr. Mohsen Sotoudeh: Eine der größten Herausforderungen ist die Balance zwischen Energiekapazität, Lebensdauer und Sicherheit der Batterien. Wir müssen Materialien finden, die nicht nur eine hohe Energiedichte ermöglichen, sondern auch stabil, sicher und umweltfreundlich sind. Ein weiteres wichtiges Thema ist die Nachhaltigkeit, insbesondere die Reduzierung der Abhängigkeit von seltenen oder schädlichen Materialien und die Verbesserung der Recyclingfähigkeit von Batterien. Schließlich ist die Skalierung der Produktion neuer Batterietechnologien eine Herausforderung, um sie wirtschaftlich und breit verfügbar zu machen.
Lasse Martinsen: Was inspiriert Sie außerhalb der Wissenschaft? Haben Sie Hobbys oder Interessen, die Ihnen neue Perspektiven oder eine Pause vom Laboralltag bieten?
Dr. Mohsen Sotoudeh: Außerhalb des Labors finde ich Entspannung und Inspiration beim mittäglichen Spaziergang mit den Kollegen. Es gibt mir die Gelegenheit, abzuschalten und gleichzeitig über meine Arbeit aus einer anderen Perspektive nachzudenken. Schwimmen ist ebenfalls ein wichtiger Bestandteil meines Lebens. Diese sportliche Aktivität hilft mir nicht nur, mich zu entspannen, sondern fördert auch meine Kreativität und Problemlösungsfähigkeiten, die ich in meine wissenschaftliche Arbeit einbringen kann.
Lasse Martinsen: Zum Abschluss, Dr. Sotoudeh, was motiviert Sie persönlich in Ihrer Arbeit, und was hoffen Sie für die Zukunft der Batterietechnologie?
Dr. Mohsen Sotoudeh: Meine größte Motivation ist die Aussicht, einen Beitrag zu einer nachhaltigeren und energieeffizienteren Zukunft zu leisten. Batterietechnologie ist ein Schlüsselbereich, der die Art und Weise, wie wir Energie speichern und nutzen, revolutionieren kann. Ich hoffe, dass wir durch unsere Forschung zu effizienteren, sichereren und umweltfreundlicheren Batteriesystemen beitragen können. Diese Systeme könnten nicht nur die Mobilität verändern, sondern auch den Zugang zu sauberer Energie weltweit verbessern. Letztendlich träume ich von einer Welt, in der saubere Energie für alle zugänglich ist und wissenschaftliche Innovationen Hand in Hand mit dem Schutz unseres Planeten gehen.
Lasse Martinsen: Das ist eine inspirierende Vision, Dr. Sotoudeh. Vielen Dank, dass Sie Ihre Einsichten und Hoffnungen mit uns geteilt haben. Ihre Arbeit und Ihr Engagement für die Wissenschaft und eine bessere Zukunft sind wirklich bewundernswert. Wir wünschen Ihnen weiterhin viel Erfolg bei Ihren Forschungen.
Dr. Mohsen Sotoudeh: Vielen Dank, Herr Martinsen. Es war mir eine Freude, hier zu sein und über meine Leidenschaft zu sprechen. Ich hoffe, dass unsere Gespräche dazu beitragen, mehr Menschen für die Wichtigkeit der Wissenschaft und insbesondere für die Entwicklungen im Bereich der Batterietechnologie zu sensibilisieren. Lassen Sie uns gemeinsam auf eine nachhaltigere Zukunft hinarbeiten.

Zukunft der Batterietechnologie: Prof. Dr. Axel Groß erörtert die Rolle der Quantenchemie bei der Materialentwicklung auf dem Batterieforum Deutschland 2024
Vom 24. bis 26. Januar 2024 fand der jährliche Kongress Batterieforum Deutschland in Berlin statt. Wissenschaftler aus Forschungs- und Entwicklungseinrichtungen sowie der Industrie vernetzten sich und tauschten sich aus. Die Schwerpunkte des dreitägigen Kongresses waren in diesem Jahr Methoden zur Beschleunigung der Zellentwicklung, Batteriezellkomponenten, Simulation und Machine Learning, Second Use, Repair und Remanufacturing sowie Batteriesysteme.
Prof. Dr. Axel Groß, Leiter des Instituts für Theoretische Chemie an der Universität Ulm, hat mit seinem eingeladenen Vortrag (Mögliche) Beiträge der Quantenchemie zur beschleunigten Materialentwicklung bei Batterien einen wichtigen Beitrag für die Diskussion zu aktuellen wissenschaftlich-technischen Schwerpunkten geliefert. In der Präsentation wurde die Verbesserung des Verständnisses von atomaren Prozessen an Oberflächen, Grenzflächen und in Batteriematerialien betont, die auf der theoretischen Beschreibung dieser Prozesse, basierend auf den Grundprinzipien der Quantenchemie, beruht und in der Lage ist Strukturen und Prozesse in Batterien durch mikroskopische Mechanismen aufzuklären.
Für das Fachpublikum war insbesondere der Ansatz zur Nutzung von Oxidperowskiten als Kathodenmaterial spannend, da diese einen Weg zur Nutzung weniger kritischer Materialien als in Lithium-Ionen-Batterien aufzeigen und somit einen Ansatz zur Entwicklung relevanter alternativer Batterietechnologien ermöglichen.
„Das Zusammentreffen von führenden Köpfen aus Wissenschaft und Industrie ist das Herzstück unseres Fortschritts", betonte Prof. Dr. Axel Groß. „Diese Art des Austausches ist unerlässlich, um innovative Lösungen für die Herausforderungen unserer Zeit zu finden. Ich bin angetan von dem Interesse und Engagement des Publikums hier auf dem Batterieforum. Nur als Gemeinschaft können wir den Weg für zukunftsweisende Technologien ebnen."
In der Tat bildet das Batterieforum Deutschland eine zentrale Drehscheibe, die weit über die Präsentation neuer Forschungsarbeiten hinausgeht. Die Veranstaltung fördert den kritischen Dialog und kreative Kollaborationen, die essenziell sind, um die Grenzen der Batterietechnologie zu erweitern. Durch die Verschmelzung verschiedenster Expertisen entwickelt sich das Forum zu einer Quelle der Inspiration, die den Teilnehmenden ermöglicht, ihre Ideen nicht nur zu teilen, sondern auch weiterzuentwickeln und in konkrete, interdisziplinäre Vorhaben zu transformieren.
Der Kongress, der nun im zwölften Jahr durchgeführt wurde, stellt eine ideale Plattform für den umfassenden Austausch über Batterietechnologie dar – er deckt das Spektrum von wissenschaftlichen Erkenntnissen, über wirtschaftliche Aspekte bis hin zu politischen Perspektiven ab. Neben etwa 300 Teilnehmern vor Ort in Berlin gab es weitere Zuhörer, die sich online dazugeschaltet hatten. Organisiert wurde die Tagung vom Kompetenznetzwerk Lithium-Ionen-Batterien (KLiB) mit Unterstützung des Bundesministeriums für Bildung und Forschung (BMBF).
Die richtungsweisenden Forschungsarbeiten des Instituts für Theoretische Chemie an der Universität Ulm erhalten maßgebliche Unterstützung durch die Dr. Barbara Mez-Starck Stiftung. Diese Förderung ermöglicht es dem Institut, an der Spitze der wissenschaftlichen Untersuchung und Entwicklung im Bereich der elektrochemischen Energiespeicherung zu stehen und dadurch wesentliche Fortschritte in der beschleunigten Materialentwicklung für Batterietechnologien zu erzielen.
Autor
Kaja G. Martinsen


Ein Schritt in Richtung verbesserte Batterietechnologien
Wenn Magnesium (Mg) auf Chlorid trifft
In der Welt der Elektrochemie sorgt eine innovative Technologie für hohe Aufmerksamkeit: Chlorid-Ionen-Batterien (CIBs). Diese innovative Entwicklung könnte eine preisgünstige und nachhaltige Alternative liefern zu der Art und Weise, wie wir Energie speichern und nutzen. Das liegt daran, dass sie auf Elementen beruht, die sehr häufig sind und damit eine hohe Versorgungssicherheit und geringe Preise versprechen. Herzstück dieses technologischen Fortschritts ist die Interaktion zwischen Halogeniden, insbesondere Chlorid, und Metallanoden. Halogenide sind die charakteristischen Anionen in Elektrolyten und ihre Rolle in Batterien geht weit über die reine Leitfähigkeit hinaus. Sie sind maßgeblich für die Effizienz und Leistungsfähigkeit von Batterien verantwortlich, indem sie stark mit metallischen Anoden reagieren. Besonders Magnesiumanoden stehen im Fokus der aktuellen Forschung, da sie neue Möglichkeiten für die Entwicklung leistungsstarker Energiespeichersysteme bieten. Diese bedeutenden Forschungsarbeiten an der Universität Ulm werden durch die Finanzierung und Unterstützung der Dr. Barbara Merz-Starck Stiftung“ ermöglicht.
Magnesium und Chlorid: Eine vielversprechende Kombination
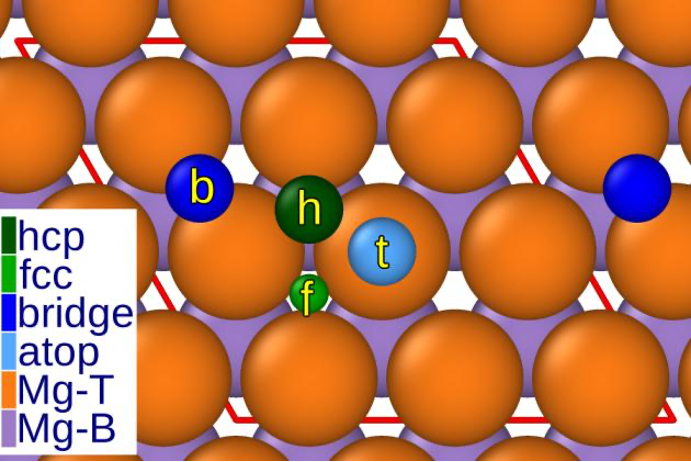
Die Wechselwirkung zwischen Mg und Chlorid zieht in der Wissenschaft große Aufmerksamkeit auf sich, insbesondere wegen der potenziellen Anwendung von Mg in CIBs. Trotz früherer Bedenken bezüglich der Korrosionsanfälligkeit von Mg durch Chlorid, haben Studien interessante Ergebnisse geliefert. Es wurde festgestellt, dass Chloridionen spezifische Stellen auf der Magnesiumoberfläche bevorzugen, besonders in hexagonal close-packed (hcp) und face-centered cubic (fcc) Hohlräumen.
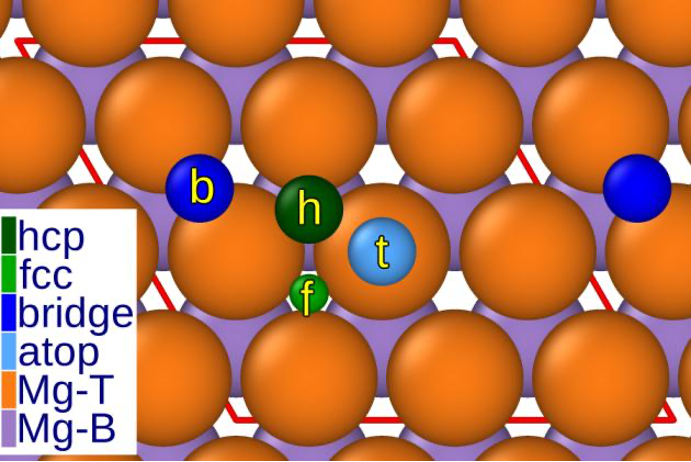
Abb. 1 Verschiedene Adsorptionsstellen auf der Magnesiumbberfläche. „Hohle“ Stelle bei hcp und fcc, die als bevorzugte Adsorptionspositionen angesehen werden können.
Salzschichten und Oberflächenstruktur
Ein weiteres Forschungsgebiet betrifft die Bildung von Salzschichten auf Metallen, ein Phänomen, das Ähnlichkeiten mit der Rostbildung aufweist. Diese Salzschichten könnten die Leistung von Magnesiumbatterien beeinflussen. Die Studie untersucht, wie Chlor (Cl) sich an Mg anheftet, ein Material, das sich durch eine stabile Oberfläche auszeichnet. Interessanterweise verändert sich der Abstand zwischen den Atomlagen auf der Magnesiumoberfläche in einer Weise, die von den meisten Metallen abweicht.
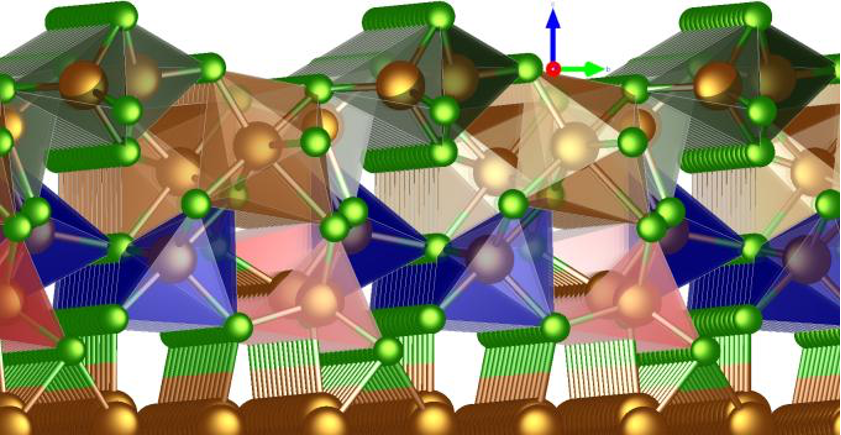
Abb. 2 Illustration der stabilsten Cl-Adsorbatstruktur
Neue Perspektiven durch Salzschichtforschung
Durch den Einsatz von Techniken wie dem "adaptive random mutation hill climbing", basierend auf Evolutionary Computing, konnten Wissenschaftler des Instituts für Theoretische Chemie der Universität Ulm die optimale Anordnung von Chlor- und Magnesiumatomen bei höheren Chlorbedeckungen bestimmen. Diese Untersuchungen offenbaren, dass mit zunehmender Chloratomanzahl die Oberflächenstruktur von Mg sich signifikant verändert, was zur Bildung einer offeneren und komplexeren Struktur führen könnte. Dieses Wissen ist entscheidend für das Verständnis der Stabilität von Magnesiumoberflächen.
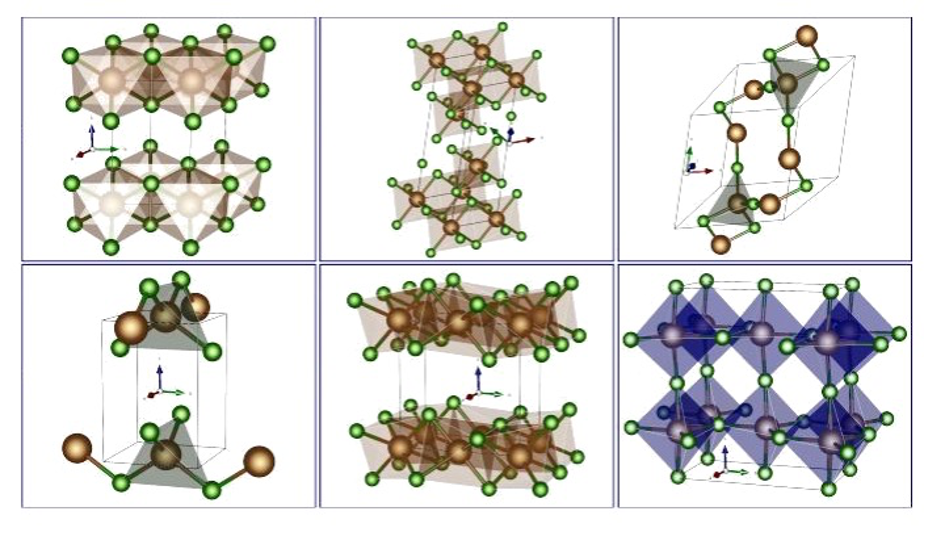
Abb. 3 Illustration unterschiedlich stabiler MgCl2 Salzstrukturen
Neue Erkenntnisse zur Salzschichtbildung auf Mg
Die eingehende Untersuchung der Salzschicht durch den Ulmer Professor Dr. Axel Groß und sein Team führte zu der Erkenntnis, dass diese Schicht aus einer Vielzahl verschiedener Magnesiumchlorid-Strukturen besteht. Dieses Ergebnis ist besonders aufschlussreich, da es anzeigt, dass die Magnesiumoberfläche durch das Hinzufügen von Chloridionen eine direkte Veränderung erfährt – ein entscheidender Vorgang in Batterien während der Energieabgabe.
Auswirkungen auf zukünftige Batterietechnologien
Die Entdeckung, dass Chloridionen die Bildung einer Salzschicht auf der Magnesiumoberfläche direkt induzieren, stellt einen innovativen Durchbruch dar. Diese Erkenntnis könnte einen wesentlichen Beitrag zur Verbesserung und Effizienzsteigerung zukünftiger Batterietechnologien leisten. Die von der Universität Ulm vorgestellten Forschungsergebnisse erweitern das Feld der Energiespeichertechnologien und legen den Grundstein für weiterführende Studien, die das Ziel verfolgen, nachhaltigere und höher performante Speicherlösungen zu entwickeln.
Quelle
1. Sarkar K, Hübner D, Stottmeister D, Gross A. Unraveling the Intricacies of Surface Salt Formation on Mg(0001): Implications for Chloride-Ion Batteries. ChemRxiv. 2023; doi:10.26434/chemrxiv-2023-hs8t9
This content is a preprint and has not been peer-reviewed.
Autor
Lasse S. Martinsen

Oxidperowskite – Pioniermaterialien für die Batterietechnologie der Zukunft?
Die Universität Ulm ebnet den Weg für nachhaltige Elektromobilität mit innovativen Post-Lithium-Batterielösungen
Die Elektrifizierung des Verkehrssektors ist ein wesentlicher Schritt, um die Klimaziele zu erreichen. Momentan dominieren Lithium-Ionen-Batterien die Technologie in Elektrofahrzeugen, da sie eine hohe Energiedichte und Leistungsfähigkeit bieten. Allerdings stehen sie vor Herausforderungen wie einer steigenden Nachfrage, die zu einer zunehmenden Verknappung führt, und Umweltbelastungen, die aus der Gewinnung und Verarbeitung dieser Materialien resultieren. Deshalb wird intensiv an alternativen Batterietechnologien geforscht, die auf weniger kritischen Materialien basieren. Ein vielversprechender Ansatz ist die Nutzung von Oxidperowskiten als Kathodenmaterial. Oxidperowskite sind Materialien mit einer charakteristischen Kristallstruktur, die sich durch Flexibilität und gute Verarbeitbarkeit auszeichnen. Sie können mit verschiedenen Metallionen dotiert werden, um die gewünschten Eigenschaften zu erreichen.
Oxidperowskite als Alternative
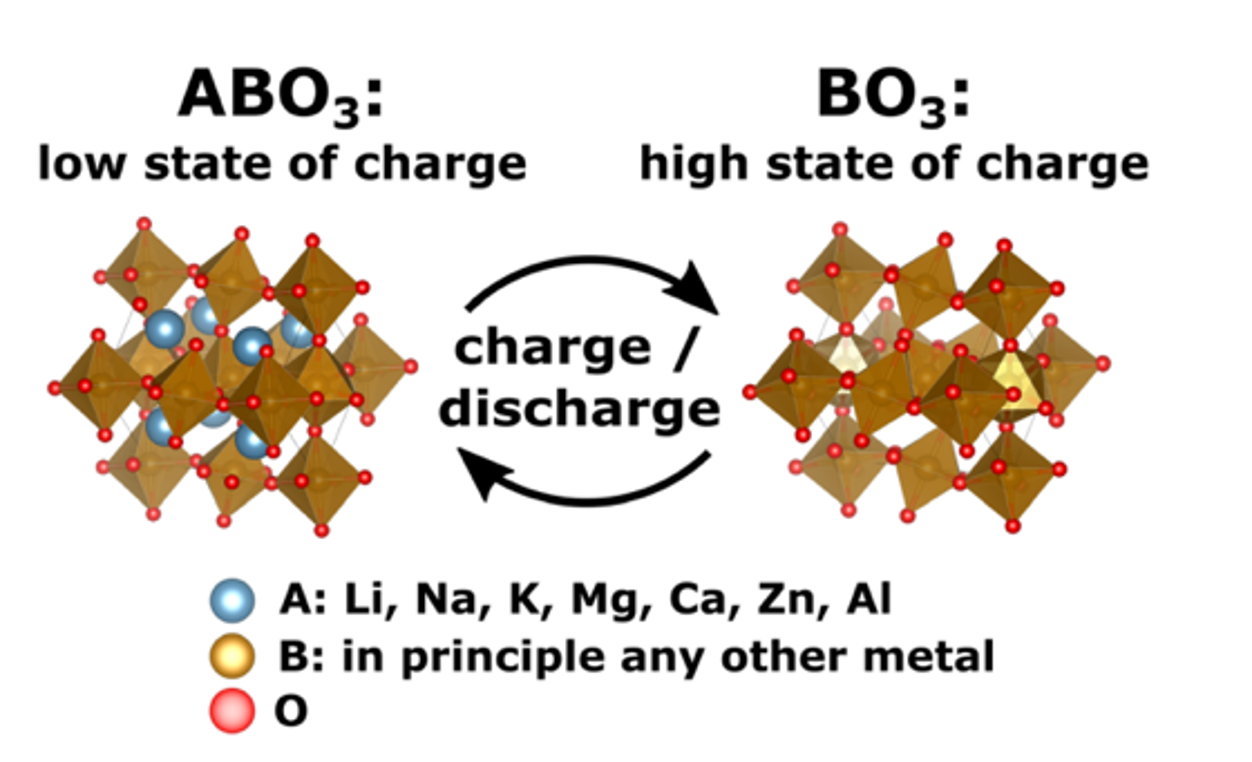
Oxidperowskite als Kathodenmaterial für Post-Lithium-Batterien stellen eine aussichtsreiche Alternative zu Lithium-Ionen-Batterien dar, da ihre Synthese unkompliziert ist. Zudem weisen sie hohe spezifische Energien sowie Energiedichten auf und gelten im Allgemeinen als umweltfreundlicher. Im Rahmen einer rechnergestützten Studie der Universität Ulm, geleitet von M.Sc. Johannes Döhn und Prof. Dr. Axel Groß, wurden 280 verschiedene Oxidperowskite hinsichtlich ihrer Tauglichkeit als Kathodenmaterial für Post-Lithium-Batterien untersucht. Unter Anwendung der Dichtefunktionaltheorie (DFT) erfolgte die Berechnung der spezifischen Energie, der Energiedichte sowie weiterer relevanter Eigenschaften dieser Verbindungen. Von den 280 Verbindungen haben 30 den ersten Screening-Prozess bestanden. Weitere 17 wurden verworfen, da u.a. die Perowskitstruktur während der DFT-Geometrieoptimierung kollabierte, so dass nur 13 Verbindungen übrig blieben. Abbildung 1 zeigt ein Kathodenmaterial für Batterien, welches auf einer Perowskit-Struktur basiert, wie sie in der vorliegenden Untersuchung analysiert wurde. Das Material ABO3 repräsentiert dabei den Zustand bei geringer Ladung und besteht aus einer vollständigen Einlagerung von Bestandteilen (hier in blau dargestellt) in das Gitter. Im Gegensatz dazu zeigt BO3 das Material im geladenen Zustand, charakterisiert durch die Entfernung dieser Bestandteile.
Die Untersuchung der verbleibenden 13 Verbindungen basierte auf den Diffusionsbarrieren für die Migration der Shuttle-Ionen, einem entscheidenden Faktor für die Batterieleistung. Zum Beispiel ist die schlechtere Ionen-Migration bei tiefen Temperaturen die Hauptursache für die geringere Reichweite von batteriegetriebenen Fahrzeugen im Winter. Die ausgewählten Materialien und ihre spezifischen Diffusionswege sind in Abbildung 2 dargestellt, welche auch die Diffusionsbarrieren der 13 erfolgreich gescreenten Verbindungen zeigt. Es ist besonders bemerkenswert, dass MgNbO3, ZnVO3 und AlMoO3 niedrige Werte an der oberen Leerstellengrenze aufzeigen, was auf eine hohe Ionenmobilität schließen lässt.
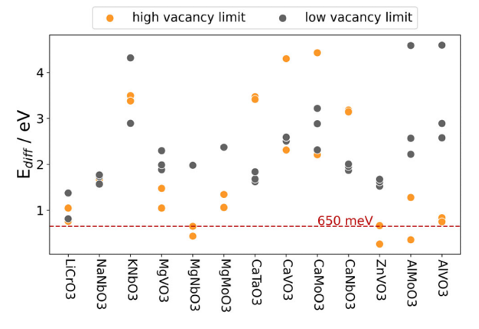
Die endgültige Auswahl umfasste MgNbO3, ZnVO3 und AlMoO3, wobei MgNbO3 besonders hervorstach.
MgNbO3: Der vielversprechendste Kandidat
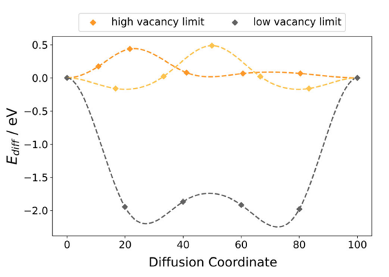
MgNbO3 wurde als besonders aussichtsreicher Kandidat für Anwendungen identifiziert. Abbildung 3 liefert eine eingehende Analyse des Diffusionsweges von MgNbO3, wobei der Diffusionspfad abwärts im interkalierten Material verläuft. Dies deutet auf eine energetisch günstige Zwischenkonfiguration statt einer endgültigen Konfiguration hin. Die Untersuchung einzelner Strukturbilder zeigt, dass die charakteristischen eckenverknüpften Sauerstoffoktaeder der Perowskitstruktur durchgehend erhalten bleiben, was die strukturelle Integrität sichert und keinen Kollaps der Struktur nahelegt. Daraus lässt sich schlussfolgern, dass die effektive Energiebarriere für die Festkörperdiffusion potenziell niedriger ist als ursprünglich angenommen und sich die Teilchen somit leichter bewegen können als gedacht.
Alltagsbezug
Die Ergebnisse der Studie, die von der MEZ Starck Stiftung mitfinanziert wurden, haben das Potenzial, die Alltagsmobilität signifikant zu beeinflussen. Durch die Verbesserung der Reichweite und Leistung von Elektrofahrzeugen könnten diese Technologien dazu beitragen, die Betriebskosten zu senken. In der Folge würden Elektrofahrzeuge für eine breitere Käuferschicht erschwinglich, was einen positiven Effekt auf die Reduktion von CO2-Emissionen haben könnte. Auf individueller Ebene bedeutet der Fortschritt, dass Konsumenten Zugang zu Elektrofahrzeugen mit besserer Leistung und größerer Reichweite haben könnten, wodurch Elektrofahrzeuge zu einer praktikableren Alternative für den persönlichen Transport avancieren. Gesellschaftlich gesehen könnte die Einführung dieser neuen Batterietechnologie die Abhängigkeit von Lithium und anderen kritischen Rohstoffen verringern, was wiederum die umweltschädlichen Auswirkungen ihrer Förderung und Verarbeitung reduzieren würde. Kurz gesagt, die Verwendung von Oxidperowskiten in Batterien könnte weitreichende positive Konsequenzen für Einzelpersonen und die Gesellschaft als Ganzes haben, von finanziellen Einsparungen bis hin zum Umweltschutz.
Ein Blick in die Zukunft
Die Forschungsergebnisse der Universität Ulm markieren einen wesentlichen Fortschritt in der Entwicklung von Post-Lithium-Batterien, die nachhaltiger und effizienter sind. Die Studie legt den Grundstein für die Optimierung der Synthese von MgNbO3, einem aussichtsreichen Material für künftige Batterietechnologien. Um die in der Theorie ermittelten Eigenschaften auch in der Praxis bestätigen zu können, bedarf es weiterer Forschungsarbeit. Zukünftige Studien in Zusammenarbeit mit der MEZ Starck Stiftung, könnten sich nicht nur auf die Verbesserung der Herstellung von MgNbO3 konzentrieren, sondern auch auf die Erhöhung der Langlebigkeit von Oxidperowskit-basierten Batterien. Darüber hinaus besteht das Ziel, neue Oxidperowskit-Verbindungen zu entwickeln, die noch höhere spezifische Energien und Energiedichten bieten.
Das sich rasant entwickelnde Gebiet der Oxidperowskit-Forschung könnte in den kommenden Jahren entscheidend zur Realisierung von Batterien beitragen, die nicht nur leistungsfähiger und langlebiger sind, sondern auch eine geringere Umweltbelastung mit sich bringen. Die Vorteile von Oxidperowskiten, wie hohe spezifische Energie und Energiedichte, prädestinieren sie als Schlüsselmaterial für die nachhaltige Batterietechnologie der Zukunft. Die aktuellen Forschungsergebnisse bilden dabei eine solide Basis für den kommerziellen Einsatz von Oxidperowskit-Batterien, doch die weitere Forschung wird entscheidend sein, um das volle Potenzial dieses Materials auszuschöpfen und in realen Anwendungen zu bestätigen.
Quelle:
Johannes Döhn and Axel Groß, Computational Screening of Oxide Perovskites as Insertion-Type Cathode Material, Adv. Energy Sustainability Res. 2300204 (2023), Open Access, DOI: 10.1002/aesr.202300204, posted on ChemRxiv, DOI: 10.26434/chemrxiv-2023-vj973 [Paper-Link]
Autor:
Lasse S. Martinsen

Unsere neuen Beiräte
Auf der letzten Vorstandsitzung der Dr. Barbara Mez-Starck-Stiftung wurden am
24. November 2023 zwei neue Beiräte ernannt. Sie werden mit uns gemeinsam in Zukunft alle Themen der Stiftung mit voranbringen und den Vorstand entsprechend unterstützen.
Frau Prof. Dr. Melanie Schnell
Professorin für Physikalische Chemie
Deutsches Elektronen-Synchrotron DESY Hamburg
sowie Universität zu Kiel
Hamburg
Herr Volker Herrdum-Heinrich
Bankdirektor (außer Dienst)
Freiburg

Verleihung des lokalen Dr. Barbara Mez-Starck-Preises an die besten Master-Absolventen
Am 19. Oktober 2023 wurden im Rahmen eines Festkolloquiums die lokalen Dr. Barbara Mez-Starck-Preise für die besten Chemie-Master-Abschlüsse des Studienjahres 2021/2022 übergeben. Vier hervorragende Master-Absolventen in Chemie wurden ausgezeichnet.

Verleihung der Ehrensenatoren-Würde
Am vergangenen Freitag, 7. Juli 2023 wurden zwei Vorstände unserer Dr. Barbara Mez-Starck Stiftung, im Rahmen des Festaktes zum 56. Jahrestag der Universität Ulm, die Ehrensenatoren-Würde verliehen. Werner Braun und Thomas Vetter sind zu Recht stolz auf diese Anerkennung für ihre Verdienste im Rahmen der langjährigen Stiftungstätigkeit.
Unsere Stiftung hat über viele Jahre hinweg die Universität Ulm und insbesondere den Ausbau des universitären Schwerpunktes „Energiewandlung und -speicherung“ unterstützt. Wir haben u.a. dabei geholfen, die Chemieinformationssysteme zu modernisieren und die eigens von Dr. Barbara Mez-Starck ins Leben gerufene MOGADOC-Datenbank weiter auszubauen, zu pflegen und weiterzuentwickeln. Sie genießt weltweites Renommee und ist heute nach wie vor ein unverzichtbares wissenschaftliches Werkzeug in der chemischen Forschung.
Ein nicht unerheblicher Teil unseres Stiftungskapitals wurde in den Bau des „Dr. Barbara Mez-Starck-Haus“ investiert, welches im Herbst 2022 zur weiteren Nutzung an die Universität Ulm übergeben wurde.
„Mit strategischem Blick haben Werner Braun und Thomas Vetter als maßgebliche Kräfte zur Neuorientierung der Stiftung beigetragen“, so Präsident Weber in seiner Laudatio. „Sie haben sich massiv dafür eingesetzt, dass der Bezug zu Dr. Barbara Mez-Starck an dieser Universität weiterhin so stark bleibt und letztlich die Nachhaltigkeit der Verbindung zwischen der Stiftung und der Universität Ulm für die Zukunft gesichert.“
Das macht uns wahrlich stolz und wir sind froh, so einen guten Partner an unserer Seite zu wissen. In Zukunft wird es noch viele neue Projekte in der kooperativen Zusammenarbeit geben, die wir als Stiftung fördern, um der Gesellschaft und insbesondere den Studierenden aus dem Fachbereich Chemie und Physik etwas zurückzugeben.

Neues Gebäude dank Stiftungsengagement –
Barbara Mez-Starck-Haus an der Uni Ulm eingeweiht
Die Barbara Mez-Starck-Stiftung hat am 27. Oktober 2022 ihr neues Büro- und Seminargebäude am Oberberghof eröffnet. Das ressourcen- und energieeffiziente Barbara Mez-Starck-Haus ist in nur eineinhalb Jahren Bauzeit fertiggestellt worden und kostete rund 6,4 Millionen Euro. Einziehen werden verschiedene Institute und Einrichtungen der Universität Ulm, die das Gebäude von der Stiftung mietet.

Am 29. August 2021 erhielten Professor Doktor Melanie Schnell (Deutsches Elektronen Synchrotron in Hamburg) und Professor Doktor Stephan Schlemmer (Universität zu Köln) den internationalen Doktor Barbara Mez-Starck-Preis.
Dr. Barbara Mez-Starck-Stiftung
Commerzbank AG
Nachlass- und Stiftungsmanagement
Kaiserstr. 16
60311 Frankfurt am Main
Telefon +49 (0) 69 – 13653934
Telefax +49 (0) 69 – 13650269
PC-Fax +49 (0) 69 – 405650056
E-Mail: info[at]mez-starck-stiftung.de